Д. Замещение в ароматическом кольце
Рассматривая реакции замещения и присоединения, мы отмечали, что алкены реагируют путем присоединения заместителей к двойной связи. Ароматические соединения вследствие резонансной стабилизации в основном вступают в реакции замещения, а не присоединения. В результате такой реакции водород замещается другой химической группой, причем наиболее часто заместителями служат группы -NO2, - Br, -Cl, -CH2CH3, -SO3H и -C(CH3)3.
Ароматическое замещение обычно происходит в две стадии. Вначале электрофильная группа атакует π-электронное облако ароматического кольца, которое затем перегруппировывается в σ-связанный аддукт с образованием sp3-гибридизованной орбитали у этого атома углерода. Это нарушает резонансную ароматическую структуру, и кольцо приобретает положительный заряд. Используя в качестве примера электрофильного ароматического замещения реакцию бромирования, отметим, что вначале бром должен вступить во взаимодействие с катализатором (обычно представляющим собой кислоту Льюиса), например с бромистым железом. В результате образуется положительно заряженный ион бромония, являющийся электрофилом
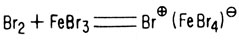
Ароматические системы обладают характерными π-электронными облаками, способными образовывать комплексы с реакционноспособными электрофильными агентами
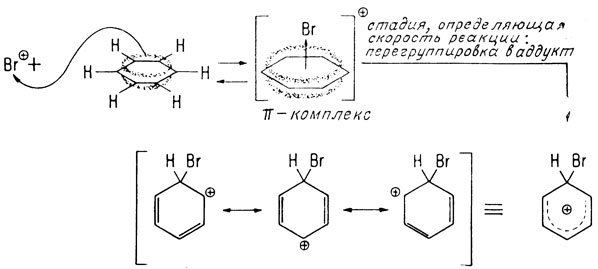
Изображение резонансной стабилизации в аддукте
Облако π-электронов бензола недостаточно реакционноспособно и поэтому образует лишь π-комплекс в концентрации, соответствующей равновесной концентрации в первой обратимой стадии. Реакции способствует лишь перегруппировка, приводящая к образованию стабилизированного σ-связью промежуточного аддукта. Во второй стадии этой реакции выделяется протон и катализатор регенерируется, а продукт замещения приобретает ароматическую структуру.

Приведенный выше пример - характерный пример реакции галогенирования ароматических соединений. Из других реакций замещения чаще всего встречаются реакции нитрования, сульфирования, алкилирования и ацилирования. Легкость, с которой идут эти реакции замещения, зависит от природы электрофильного замещающего агента и от возможности атаки им π-электронов ароматического кольца. Электрофильное замещение ароматических соединений, π-электронная структура которых выражена наиболее сильно, происходит с наибольшими скоростями.
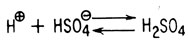
Нитрование аналогично галогенированию и другим реакциям ароматического замещения также является двухстадийным процессом, причем скорость всего процесса определяет первая стадия. Для нитрования, как и для других реакций ароматического замещения, требуется участие катализатора. Обычно для образования из азотной кислоты иона нитрония NO+2 применяют серную кислоту. В концентрированной серной кислоте (например, 92%-ной) азотная кислота существует почти полностью в виде иона нитрония. Поэтому скорость таких реакций нитрования выражается уравнением второго порядка:
Механизм нитрования в таких условиях следующий:
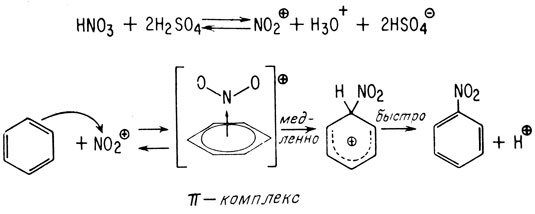
Если ионы нитрония образуются медленно, а ароматическое соединение по реакционной способности близко к бензолу, ионы нитрония будут расходоваться тотчас же по мере их образования. Такое положение наблюдается при нитровании азотной кислотой в уксусной кислоте или в нитрометане, взятых в качестве растворителей. В этом случае скорость зависит от концентрации ароматического соединения и от эффективности образования ионов нитрония. Если реакционная способность ароматического соединения меньше реакционной способности бензола (например, этиловый эфир бензойной кислоты, галогензамещенные бензолы и т. д.), скорость нитрования будет зависеть только от концентрации ароматического соединения (реакция псевдопервого порядка):
Если в ароматическое кольцо уже введена одна нитрогруппа, его π-электроны становятся менее доступными для атаки ионом нитрония. Так, толуол легко нитруется при температуре от 0 до 25°С. Получающийся в результате такого нитрования продукт нитруется в среде толуола значительно труднее, чем толуол в аналогичных условиях. Однако при достаточном нагревании может образовываться 2,4-динитротолуол и даже 2,4,6-тринитротолуол (ТНТ) (H2SO4 с SO3 - дымящая серная кислота)
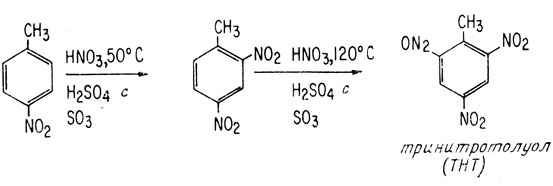
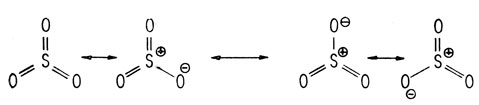
Сульфирование является примером реакции замещения нейтральным электрофильным агентом SO3. Электрофильный характер атома серы явственно виден из рассмотрения следующих резонансных гибридных состояний серного ангидрида:
Обычно для сульфирования бензола и других ароматических соединений применяют дымящую серную кислоту (H2SO4-SO3).
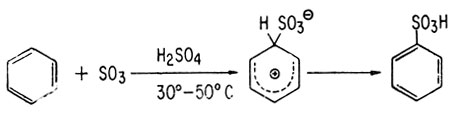
Если для сульфирования вместо дымящей серной кислоты использовать концентрированную серную кислоту, то требуются повышенные температуры. Было установлено, что реакции сульфирования серным ангидридом в нитробензоле - реакции третьего порядка:
Механизм, в основе которого лежит такое кинетическое выражение, должен включать участие комплекса димерной SO3 с ароматическим соединением на стадии, определяющей скорость реакции.
В 1887 г. французский химик Шарль Фридель и работавший у него американский студент Джеймс Крафтс открыли весьма важную для органической химии реакцию, которая в настоящее время называется реакцией алкилирования по Фриделю-Крафтсу. Используя в качестве катализатора небольшое количество галогенида металла, они присоединили алкилгалогенид к ароматическому соединению. Впоследствии, изучая эту реакцию, химики обнаружили, что алкилирование происходит в том случае, если имеется ион карбония, который атакует ароматическое кольцо. Так, вместо алкилгалогенидов можно использовать спирты и алкены. Ниже приведены примеры реакции алкилирования по Фриделю-Крафтсу:
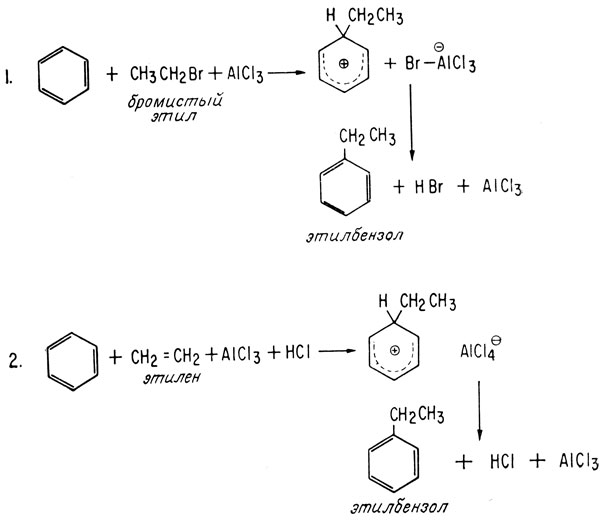
Катализатор алкилирования, хлористый алюминий, может применяться в небольших нестехиометрических количествах, поскольку в ходе реакции он регенерируется. И в этом случае продукты алкилирования бензола также обычно обладают более высокой реакционной способностью, чем сам бензол. Поэтому если хотят получить моноалкилбензол, то необходимо принимать меры для его быстрого удаления из реакционной среды с тем, чтобы предотвратить его дальнейшее алкилирование.
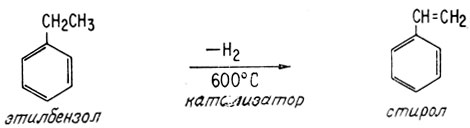
При выделении молекулы водорода из этилбензола, полученного одним из указанных выше способов под действием катализатора или при нагревании до весьма высоких температур, образуется стирол - соединение, используемое при синтезе пластических масс и синтетического каучука (гл. 4)
В случае алкилгалогенида первоначально, по-видимому, образуется первичный ион карбония, который быстро перегруппировывается в гораздо более стабильный вторичный ион карбония (реакция 3).
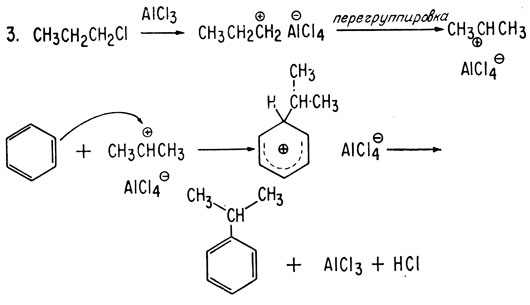
В качестве катализаторов реакции алкилирования чаще всего используют кислоты Льюиса, например хлористый алюминий (AlCl3) и трехфтористый бор (BF3), а из алкилгалогенидов обычно применяют хлор- или бромпроизводные. Несмотря на самые тщательные меры предосторожности, обычно образуются полизамещенные продукты, и для выделения определенного продукта из смеси нескольких соединений, полученных при синтезе, требуется тщательная очистка. Число образующихся при синтезе продуктов еще больше увеличивается за счет тенденции н-алкилгалогенидов образовывать заместители с различным характером разветвления (примером является приведенная выше реакция 3). Характер разветвления заместителя зависит от температуры, и особенно легко разветвление происходит при высоких температурах.
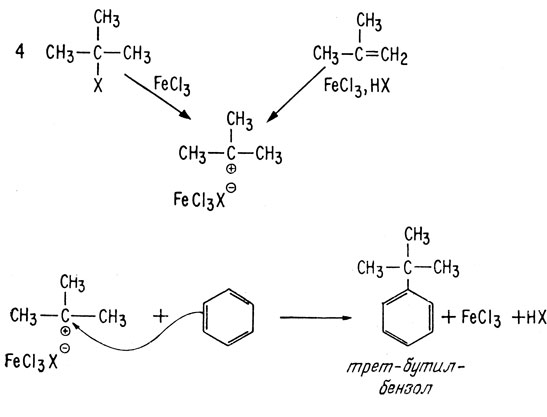
Предшественниками, из которых легко образуются электрофилы (реакция 4), служат алифатические спирты, алкилгалогениды и разветвленные алкены, способные образовывать третичные ионы карбония. Ароматические соединения могут атаковать также электрофилы, в результате чего образуются алкилароматические соединения, содержащие четвертичный замещенный атом углерода.
Другой, более специфической, чем алкилирование, является реакция ацилирования по Фриделю-Крафтсу. В результате реакций этого типа из хлорангидрида кислоты RCOC1 или ангидрида (RCO)2O и ароматического углеводорода образуется кетон. В качестве катализатора чаще всего используют AlCl3, действие которого заключается в создании электрофильных групп.
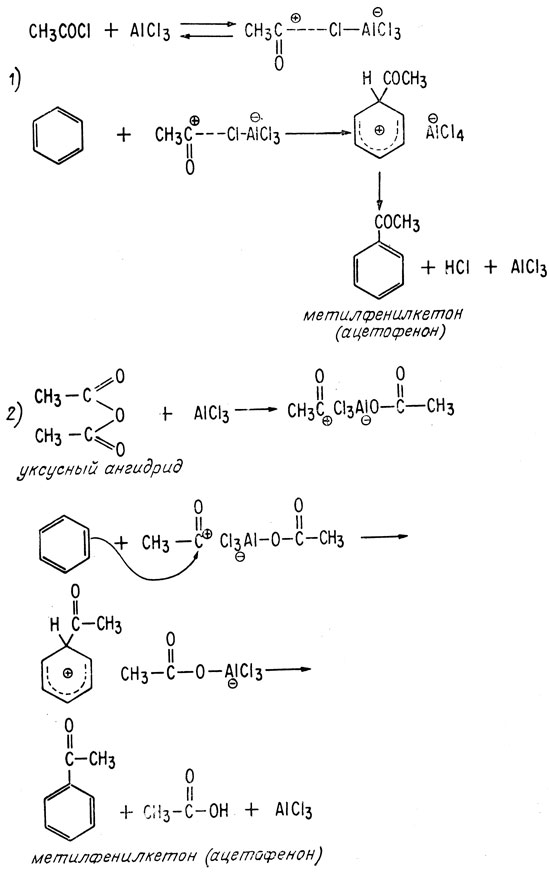
Реакции этого типа отличаются от алкилирования тем, что чаще образуется чистый продукт, а не смесь продуктов и что для ацилирования требуются большие количества катализатора, поскольку катализатор образует комплекс с получающимся кетоном.
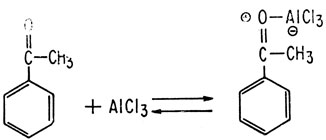
Если в качестве реагента используется хлорангидрид кислоты, необходимо брать катализатор в количестве немногим более одного молярного эквивалента; если реагентом является ангидрид, катализатор применяют в количестве, несколько превышающем два молярных эквивалента.
Присутствие в ароматическом кольце заместителей влияет на легкость замещения другими заместителями. Это влияние увеличивается при действии электронов замещающих групп на π-электроны ароматического кольца. Если атом заместителя, присоединенный к ароматическому кольцу, имеет положительный заряд или является акцептором электронов, тогда π-электроны кольца притягиваются к этому атому и вероятность их атаки на электрофил уменьшается. Примером таких заместителей, приводящих к деактивации кольца, являются -NO2, - SO3H, -Cl, сложноэфирные и кетонные группы. (Дезактивирующим действием кетонных групп объясняется получение чистых монозамещенных продуктов при ацилировании по Фриделю-Крафтсу.)
Замещение, наоборот, облегчается при наличии в ароматическом кольце электронодонорных заместителей. Такие заместители увеличивают электронную плотность в кольце, и вследствие этого атака электрофила кольцом облегчается. Примерами активирующих групп являются -NH2, -ОН, -ОСН3 и алкильные группы. Заместители влияют не только на легкость введения последующих заместителей в ароматическое кольцо, но также и на их положение в кольце. Напомним, что в одном из предыдущих разделов мы определяли названия орто, мета и пара как замещение в соседнее положение, через один атом углерода или в противоположное первому положению соответственно.
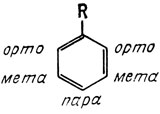
Вследствие наличия резонанса между исходным заместителем и самим кольцом увеличивается электронная плотность либо на атомах углерода, находящихся в мета-положениях, либо на атомах, находящихся в орто-пара-положениях. В качестве примера рассмотрим резонансные гибридные структуры метоксибензола:
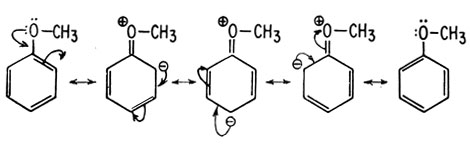
На основании такого рассмотрения можно предсказать, что введение метоксигруппы будет активировать ароматическое кольцо в реакции замещения и что новые электрофильные заместители будут вступать преимущественно в орто- и (или) пара-положение. Поэтому группу -ОСН3 называют орто-пара-ориентантом (направляет заместитель в орто-пара-положения). Большинство орто-пара-ориентантов являются активаторами (табл. 8). Исключение составляют галогены: они служат орто-пара-ориентантами, но являются дезактиваторами реакции замещения вследствии своей большой электроотрицательности.
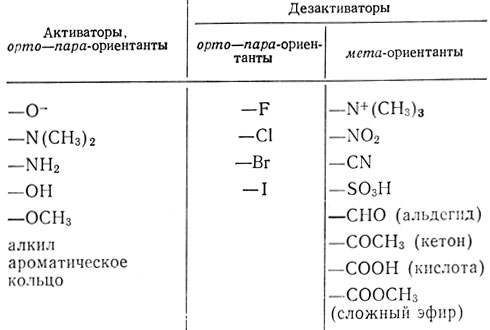
Таблица 8. Влияние заместителей в ароматическом кольце* на реакционную способность и место присоединения электрофила к кольцу
* (В порядке уменьшения активности.)
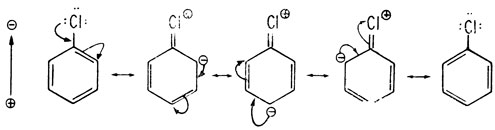
Резонансные гибридные структуры для хлорбензола
Все мета-направляющие заместители являются дезактиваторами. Хотя дезактиваторы уменьшают легкость атаки электрофила ароматическим кольцом, замещение все-таки возможно. Если замещение происходит, оно в первую очередь идет в мета-положение. Так, нитрогруппа в нитробензоле служит дезактиватором и мета-ориентантом. Она приводит к такому смещению электронов, что электронная плотность атомов углерода, находящихся в орто- и пара-положениях, становится меньше, чем у атомов углерода, находящихся в мета-положениях. Поэтому вероятность атаки электрофильной группы со стороны атомов углерода, находящихся в орто- или пара-положениях, будет меньше, чем со стороны атомов углерода, находящихся в мета-положениях. В этих условиях специфичность ориентации замещения ниже, чем при наличии орто-пара-ориентантов.
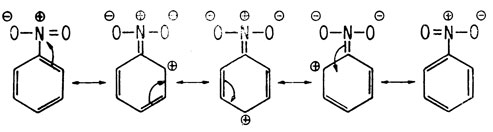
Резонансные гибридные структуры для нитробензола
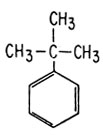
Как было показано выше, ориентация замещения определяется в первую очередь резонансом. Однако существенную роль играют и пространственные затруднения, которые объясняют, почему многие орто-пара-ориентанты на самом деле благоприятствуют замещению в пара-положение. Так, объемистая трет-бутильная группа направляет электрофильные заместители почти исключительно в пара-положение. Заместитель просто не может "протиснуться" в орто-положение, доступ к которому ему закрывает объемистая трет-бутильная группа.
Алексей Корнеев teamjet, касса | снять квартиру от собственника
|
ПОИСК:
|
При копировании ссылка обязательна:
http://biologylib.ru/ 'Библиотека по биологии'